Tutorial
Ready-to-run scripts for the functionalities introduced here can be found in the examples directory of the repo.
Creating a bacterium
Bacteria are represented by custom types, that must be subtypes of the AbstractAgent type implemented by Agents.jl.
Bactos.AbstractMicrobe — TypeAbstractMicrobe{D} <: AbstractAgent where {D<:Integer}
YourMicrobeType{D} <: AbstractMicrobe{D}All microbe types in Bactos.jl simulations must be instances of user-defined types that are subtypes of AbstractMicrobe. The parameter D defines the dimensionality of the space in which the microbe type lives (1, 2 and 3 are currently supported).
All microbe types should have the following fields: - id::Int → id of the microbe - pos::NTuple{D,Float64} → position of the microbe - vel::NTuple{D,Float64} → velocity of the microbe - motility → motile pattern of the microbe - turn_rate::Float64 → average reorientation rate of the microbe - rotational_diffusivity::Float64 → rotational diffusion coefficient
By default, Bactos.jl provides a basic Microbe type, that is usually sufficient for the simplest types of simulations.
Bactos.Microbe — TypeMicrobe{D} <: AbstractMicrobe{D}Basic microbe type for simple simulations.
Default parameters:
id::Int→ identifier used internally by Agents.jlpos::NTuple{D,Float64} = ntuple(zero,D)→ positionmotility = RunTumble()→ motile patternvel::NTuple{D,Float64} = rand_vel(D) .* rand(motility.speed)→ velocity vectorturn_rate::Float64 = 1.0→ frequency of reorientationsstate::Float64→ generic variable for a scalar internal staterotational_diffusivity::Float64 = 0.0→ rotational diffusion coefficientradius::Float64 = 0.0→ equivalent spherical radius of the microbe
In order to create a Microbe living in a one-dimensional space we can just call
Microbe{1}(id=0)It is required to pass a value to the id argument (this behavior might change in the future). All the other parameters will be given default values (as described in the type docstring) if not assigned explicitly.
Similarly, for bacteria living in two or three dimensions we can use
Microbe{2}(id=0)
Microbe{3}(id=0)Custom parameters can be set via kwargs:
Microbe{3}(
id = 0,
pos = (300.0, 0.0, 0.0),
motility = RunTumble(speed = Normal(40.0, 4.0)),
vel = rand_vel(3) .* 40.0,
turn_rate = 1.5,
state = 0.0,
rotational_diffusivity = 0.035,
radius = 0.5
)Creating a model
BacteriaBasedModel provides a fast way to initialise an AgentBasedModel (from Agents.jl) via the initialise_model function, using a typical procedure. If higher levels of customization are needed, the model will need to be created by hand.
Bactos.initialise_model — Functioninitialise_model(;
microbes,
timestep,
extent, spacing = extent/20, periodic = true,
random_positions = true,
model_properties = Dict(),
)Initialise an AgentBasedModel from population microbes. Requires the integration timestep and the extent of the simulation box.
When random_positions = true the positions assigned to microbes are ignored and new ones, extracted randomly in the simulation box, are assigned; if random_positions = false the original positions in microbes are kept.
Any extra property can be assigned to the model via the model_properties dictionary.
We can now generate a population of microbes and, after choosing an integration timestep and a domain size, we initialise our model, placing the microbes at random locations in the domain.
microbes = [Microbe{3}(id=i) for i in 1:10]
timestep = 0.1
extent = 100.0
model = initialise_model(;
microbes = microbes,
timestep = timestep,
extent = extent
)AgentBasedModel with 10 agents of type Microbe
space: periodic continuous space with (100.0, 100.0, 100.0) extent and spacing=5.0
scheduler: fastest
properties: timestepRandom walks
Now we can already generate random walks. The setup follows previous sections.
timestep = 0.1
extent = 1e6 # just a large value to stay away from boundaries
nmicrobes = 8
# initialise all microbes at same position
microbes = [Microbe{1}(id=i, pos=(L/2,)) for i in 1:nmicrobes]
model = initialise_model(;
microbes,
timestep,
extent, periodic = false,
random_positions = false
)Now we need to define the adata variable to choose what observables we want to track, throughout the simulation, for each agent in the system. In our case, only the position field
adata = [:pos]Now we can run the simulation; the microbe_step! function will take care of the stepping and reorientations according to the properties of each microbe:
nsteps = 1000
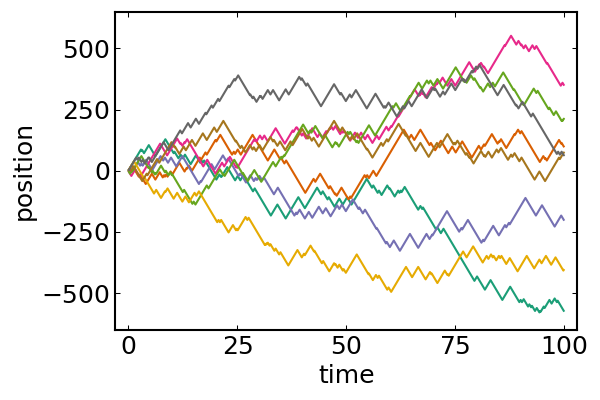
adf, = run!(model, microbe_step!, nsteps; adata)x = first.(vectorize_adf_measurement(adf, :pos))'
plot(
(0:nsteps).*dt, x,
legend = false,
xlab = "time",
ylab = "position"
)
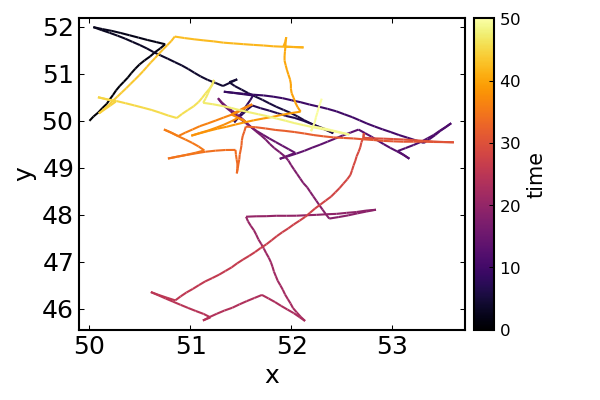
Similarly for a two-dimensional random walk, using run-reverse-flick motility and non-zero rotational diffusion:
dt = 0.1
L = 1000.0
nmicrobes = 1
microbes = [
Microbe{2}(
id=i, pos=(L/2,L/2),
motility=RunReverseFlick(),
rotational_diffusivity = 0.2,
) for i in 1:nmicrobes
]
model = initialise_model(;
microbes,
timestep = dt,
extent, periodic = false,
random_positions = false,
)
nsteps = 500
adata = [:pos]
adf, = run!(model, microbe_step!, nsteps; adata)
traj = vectorize_adf_measurement(adf, :pos)
x = first.(traj)'
y = last.(traj)'
plot(
x, y, line_z = (0:nsteps).*dt,
legend=false,
xlab = "x", ylab = "y",
colorbar = true, colorbar_title = "time"
)
Microbes with different motile patterns can also be combined in the same simulation, without extra complications or computational costs:
n = 3
microbes_runtumble = [Microbe{2}(id=i, motility=RunTumble()) for i in 1:n]
microbes_runrev = [Microbe{2}(id=n+i, motility=RunReverse()) for i in 1:n]
microbes_runrevflick = [Microbe{2}(id=2n+1, motility=RunReverseFlick()) for i in 1:n]
microbes = vcat(
microbes_runtumble, microbes_runrev, microbes_runrevflick
)Chemotaxis in a linear gradient
We will now reproduce a classical chemotaxis assay: bacteria in a rectangular channel with a linear attractant gradient.
Bactos.jl requires three functions to be defined for the built-in chemotaxis models to work: concentration_field, concentration_gradient, and concentration_time_derivative; all three need to take the two arguments (pos, model). First we need to define our concentration field and its gradient (we don't define its time derivative since it will be held constant). We will use a linear gradient in the x direction. Here we can define also the gradient analytically, in more complex cases it can be evaluated numerically through the finite difference interface.
concentration_field(x,y,C₀,∇C) = C₀ + ∇C*x
function concentration_field(pos, model)
x, y = pos
C₀ = model.C₀
∇C = model.∇C
concentration_field(x, y, C₀, ∇C)
end
concentration_gradient(x,y,C₀,∇C) = [∇C, 0.0]
function concentration_gradient(pos, model)
x, y = pos
C₀ = model.C₀
∇C = model.∇C
concentration_gradient(x, y, C₀, ∇C)
endWe choose the parameters, initialise the population (with two distinct chemotaxers) with all bacteria to the left of the channel, and setup the model providing the functions for our concentration field to the model_properties dictionary.
timestep = 0.1 # s
Lx, Ly = 1000.0, 500.0 # μm
extent = (Lx, Ly) # μm
periodic = false
n = 50
microbes_brumley = [
Brumley{2}(id=i, pos=(0,rand()*Ly), chemotactic_precision=1)
for i in 1:n
]
microbes_brown = [
BrownBerg{2}(id=n+i, pos=(0,rand()*Ly))
for i in 1:n
]
microbes = [microbes_brumley; microbes_brown]
C₀ = 0.0 # μM
∇C = 0.01 # μM/μm
model_properties = Dict(
:concentration_field => concentration_field,
:concentration_gradient => concentration_gradient,
:concentration_time_derivative => (_,_) -> 0.0,
:compound_diffusivity => 500.0, # μm²/s
:C₀ => C₀,
:∇C => ∇C,
)
model = initialise_model(;
microbes,
timestep,
extent, periodic,
model_properties,
random_positions = false
)Notice that we also defined an extra property compound_diffusivity. This quantity is required by the models of chemotaxis that use sensing noise (such as Brumley, XieNoisy, CelaniNoisy). 500 μm²/s is a typical value for small molecules.
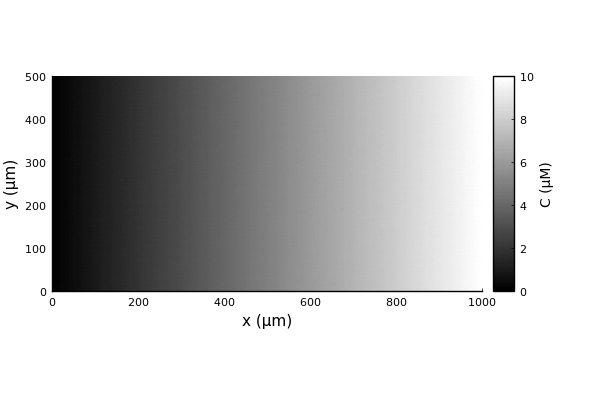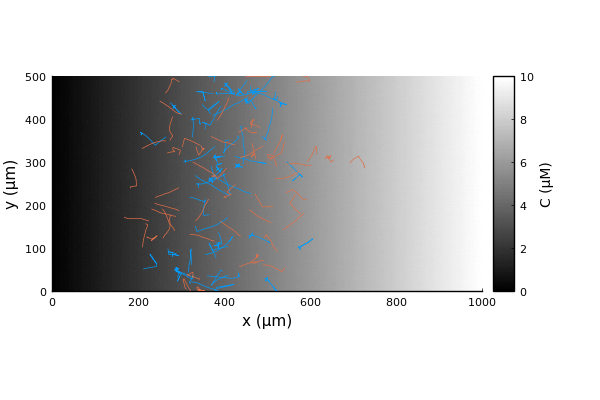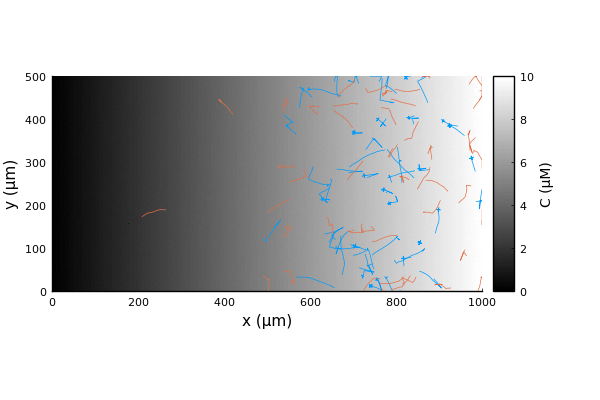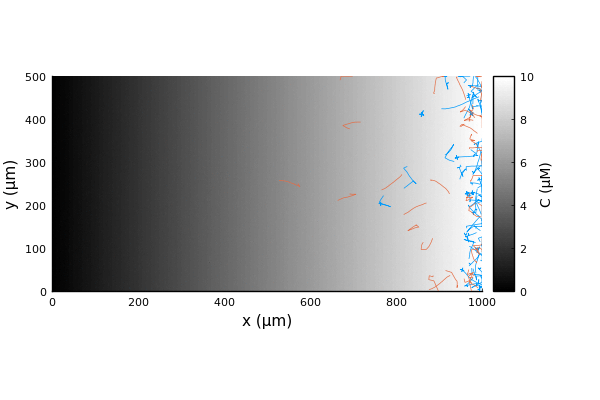
We can run the simulation as usual and extract the trajectories.
adata = [:pos]
nsteps = 1000 # corresponds to 100s
adf, = run!(model, microbe_step!, nsteps; adata)
traj = vectorize_adf_measurement(adf, :pos)
x = first.(traj)'
y = last.(traj)'Comparing the trajectories for the two bacterial species we witness a chemotactic race (Brumley in blue, BrownBerg in orange).