Sequence Types
BioSequences exports an abstract BioSequence type, and several concrete sequence types which inherit from it.
The abstract BioSequence
BioSequences provides an abstract type called a BioSequence{A<:Alphabet}. This abstract type, and the methods and traits is supports, allows for many algorithms in BioSequences to be written as generically as possible, thus reducing the amount of code to read and understand, whilst maintaining high performance when such code is compiled for a concrete BioSequence subtype. Additionally, it allows new types to be implemented that are fully compatible with the rest of BioSequences, providing that key methods or traits are defined).
This abstract type is parametric over concrete types of Alphabet, which define the range of symbols permitted in the sequence.
Some aliases are also provided for your convenience:
| Type alias | Type |
|---|---|
NucleotideSeq | BioSequence{<:NucleicAcidAlphabet} |
AminoAcidSeq | BioSequence{AminoAcidAlphabet} |
Any concrete sequence type compatible with BioSequences must inherit from BioSequence{A}, where A is the alphabet of the concrete sequence type. It must also have the following methods defined for it:
BioSequences.encoded_data — FunctionReturn the data member of seq that stores the encoded sequence data.
Base.length — MethodGet the length of a biological sequence.
If these requirements are satisfied, the following key traits and methods backing the BioSequences interface, should be defined already for the sequence type.
BioSequences.encoded_data_type — FunctionGet the vector of bits storing a sequences packed encoded elements.
BioSequences.encoded_data_eltype — FunctionGet the element type of the vector of bits storing a sequences packed encoded elements.
BioSequences.Alphabet — MethodReturn the Alpahbet type that defines the biological symbols allowed for seq.
BioSymbols.alphabet — MethodGets the alphabet encoding of a given BioSequence.
BioSequences.BitsPerSymbol — TypeThe number of bits required to represent a packed symbol in a vector of bits.
BioSequences.bits_per_symbol — FunctionGet the number of bits each symbol packed into a BioSequence uses, as an integer value.
As a result, the vast majority of methods described in the rest of this manual should work out of the box for the concrete sequence type. But they can always be overloaded if needed.
Long Sequences
Many genomics scripts and tools benefit from an efficient general purpose sequence type that allows you to create and edit sequences. In BioSequences, the LongSequence type fills this requirement.
LongSequence{A<:Alphabet} <: BioSequence{A} is parameterized by a concrete Alphabet type A that defines the domain (or set) of biological symbols permitted. For example, AminoAcidAlphabet is associated with AminoAcid and hence an object of the LongSequence{AminoAcidAlphabet} type represents a sequence of amino acids. Symbols from multiple alphabets can't be intermixed in one sequence type.
The following table summarizes common LongSequence types that have been given aliases for convenience.
| Type | Symbol type | Type alias |
|---|---|---|
LongSequence{DNAAlphabet{4}} | DNA | LongDNASeq |
LongSequence{RNAAlphabet{4}} | RNA | LongRNASeq |
LongSequence{AminoAcidAlphabet} | AminoAcid | LongAminoAcidSeq |
LongSequence{CharAlphabet} | Char | LongCharSeq |
The LongDNASeq and LongRNASeq aliases use a DNAAlphabet{4}, which means the sequence may store ambiguous nucleotides. If you are sure that nucleotide sequences store unambiguous nucleotides only, you can reduce the memory required by sequences by using a slightly different parameter: DNAAlphabet{2} is an alphabet that uses two bits per base and limits to only unambiguous nucleotide symbols (ACGT in DNA and ACGU in RNA). Replacing LongSequence{DNAAlphabet{4}} in your code with LongSequence{DNAAlphabet{2}} is all you need to do in order to benefit. Some computations that use bitwise operations will also be dramatically faster.
Kmers & Skipmers
Kmers
Bioinformatic analyses make extensive use of kmers. Kmers are contiguous sub-strings of k nucleotides of some ref sequence.
They are used extensively in bioinformatic analyses as an informational unit. This concept popularised by short read assemblers. Analyses within the kmer space benefit from a simple formulation of the sampling problem and direct in-hash comparisons.
BioSequences provides the following types to represent Kmers, unlike some sequence types, they are immutable.
Mer{A<:NucleicAcidAlphabet{2},K}
Represents a substring of K DNA or RNA nucleotides (depending on the alphabet A). This type represents the sequence using a single UInt64, and so the maximum possible sequence possible - the largest K possible, is 32. We recommend using 31 rather than 32 however, as odd K values avoid palindromic mers which can be problematic for some algorithms.
BigMer{A<:NucleicAcidAlphabet{2},K}
Represents a substring of K DNA or RNA nucleotides (depending on the alphabet A). This type represents the sequence using a single UInt128, and so the maximum possible sequence possible - the largest K possible, is 64. We recommend using 63 rather than 64 however, as odd K values avoid palindromic mers which can be problematic for some algorithms.
AbstractMer{A<:NucleicAcidAlphabet{2},K}
This abstract type is just a type that unifies the Mer and BigMer types for the purposes of writing generalised methods of functions.
Several aliases are provided for convenience:
| Type alias | Type |
|---|---|
DNAMer{K} | Mer{DNAAlphabet{2},K} |
RNAMer{K} | Mer{RNAAlphabet{2},K} |
DNAKmer | DNAMer{31} |
RNAKmer | RNAMer{31} |
BigDNAMer{K} | BigMer{DNAAlphabet{2},K} |
BigRNAMer{K} | BigMer{RNAAlphabet{2},K} |
BigDNAKmer | BigMer{DNAAlphabet{2},63} |
BigRNAKmer | BigMer{RNAAlphabet{2},63} |
DNACodon | DNAMer{3} |
RNACodon | RNAMer{3} |
Skipmers
For some analyses, the contiguous nature of kmers imposes limitations. A single base difference, due to real biological variation or a sequencing error, affects all k-mers crossing that position thus impeding direct analyses by identity. Also, given the strong interdependence of local sequence, contiguous sections capture less information about genome structure, and so they are more affected by sequence repetition.
Skipmers are a generalisation of the concept of a kmer. They are created using a cyclic pattern of used-and-skipped positions which achieves increased entropy and tolerance to nucleotide substitution differences by following some simple rules.
Skipmers preserve many of the elegant properties of kmers such as reverse complementability and existence of a canonical representation. Also, using cycles of three greatly increases the power of direct intersection between the genomes of different organisms by grouping together the more conserved nucleotides of protein-coding regions.
BioSequences currently does not provide a separate type for skipmers, they are represented using Mer and BigMer as their representation as a short immutable sequence encoded in an unsigned integer is the same. The distinction lies in how they are generated.
Skipmer generation
A skipmer is a simple cyclic q-gram that includes m out of every n bases until a total of k bases is reached.
This is illustrated in the figure below (from this paper.):
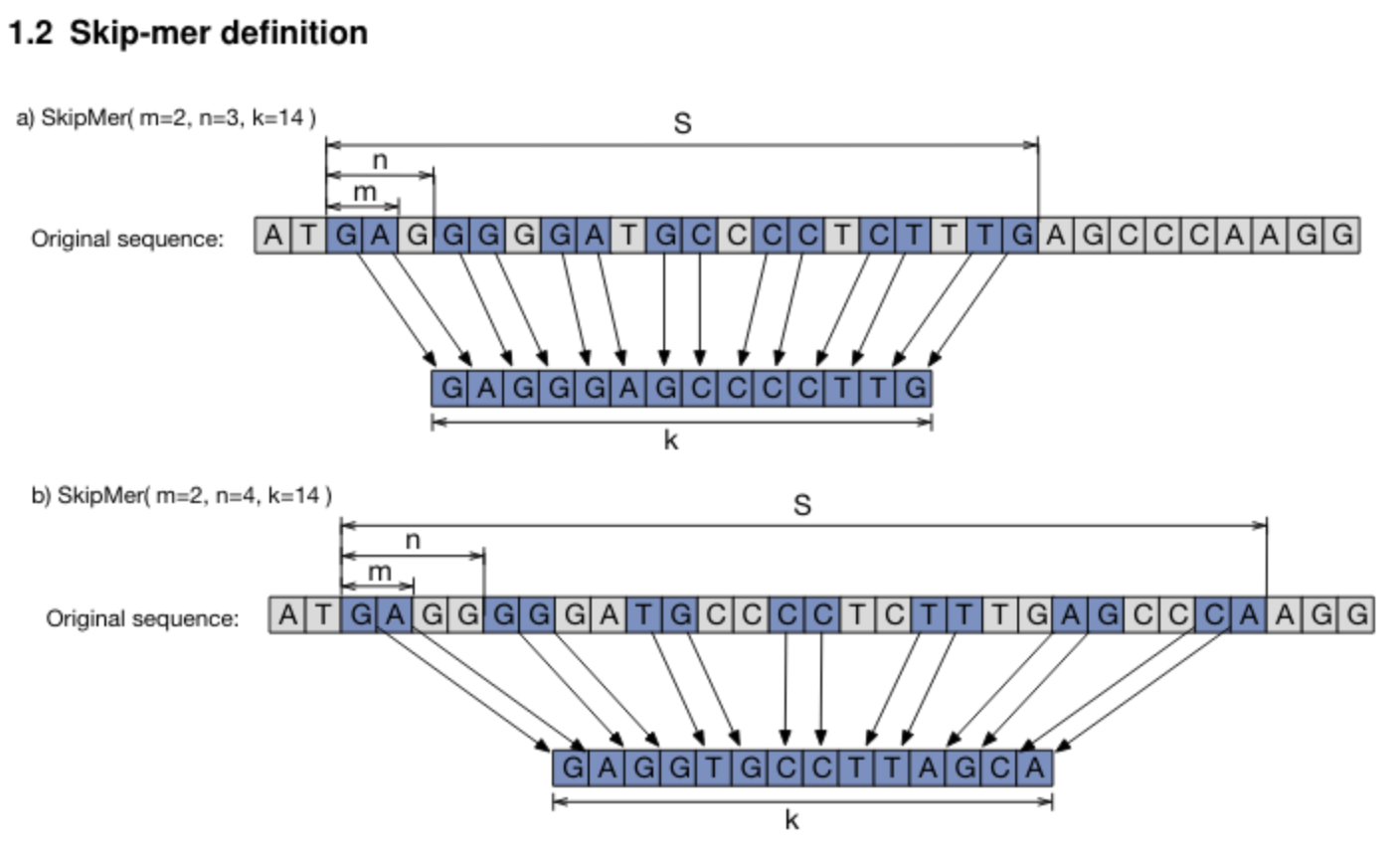
To maintain cyclic properties and the existence of the reverse-complement as a skipmer defined by the same function, k should be a multiple of m.
This also enables the existence of a canonical representation for each skipmer, defined as the lexicographically smaller of the forward and reverse-complement representations.
Defining m, n and k fixes a value for S, the total span of the skipmer, given by:
To see how to iterate over skipmers cf. kmers, see the Iteration section of the manual.
Reference sequences
LongDNASeq (alias of BioSequence{DNAAlphabet{4}}) is a flexible data structure but always consumes 4 bits per base, which will waste a large part of the memory space when storing reference genome sequences. In such a case, ReferenceSequence is helpful because it compresses positions of 'N' symbols so that long DNA sequences are stored with almost 2 bits per base. An important limitation is that the ReferenceSequence type is immutable due to the compression. Other sequence-like operations are supported:
julia> seq = ReferenceSequence(dna"NNCGTATTTTCN")
12nt Reference Sequence:
NNCGTATTTTCN
julia> seq[1]
DNA_N
julia> seq[5]
DNA_T
julia> seq[2:6]
5nt Reference Sequence:
NCGTA
julia> ReferenceSequence(dna"ATGM") # DNA_M is not accepted
ERROR: ArgumentError: invalid symbol M ∉ {A,C,G,T,N} at 4
in convert at /Users/kenta/.julia/v0.4/Bio/src/seq/refseq.jl:58
in call at essentials.jl:56
Alphabet types
BioSequences.Alphabet — TypeAlphabets of biological symbols.
Alphabet is perhaps the most important type trait for biological sequences in BioSequences.jl.
An Alphabet represents a domain of biological symbols.
For example, DNAAlphabet{2} has a domain of unambiguous nucleotides (i.e. A, C, G, and T).
Alphabet types restrict and define the set of biological symbols, that can be encoded in a given biological sequence type. They ALSO define HOW that encoding is done.
An Alphabet type defines the encoding of biological symbols with a pair of associated encoder and decoder methods. These paired methods map between biological symbol values and a binary representation of the symbol.
Any type A <: Alphabet, is expected to implement the Base.eltype method for itself. It is also expected to implement the BitsPerSymbol method.
Alphabets control how biological symbols are encoded and decoded. They also confer many of the automatic traits and methods that any subtype of T<:BioSequence{A<:Alphabet} will get.